RESEARCH PROJECTS
1. Lighter and stronger materials for extreme environments
In this work we focus on developing a new class of magnesium alloys for extreme dynamic environments. Materials under dynamic environment fail due to spall, and the mechanism for this is primarily due to the nucleation of nanovoids from high tensile stresses, leading to void growth and coalescence, and eventually, the propagation of cracks and material failure. In my research, I have worked on developing lightweight materials with higher spall resistance. I have specifically focused on magnesium and its alloys which are attractive and achieve desirable mechanical properties via age hardening; and precipitates of magnesium-aluminum is important in imparting spall strength and existing alloys of magnesium have low spall strength. To this end, I have worked on understanding the influence of stresses and strains (which arise during processing) on the precipitation process in magnesium-aluminum alloys. I have used DFT to accurately calculate the energetics of binary magnesium-aluminum solid solutions. Our results show that the energetics of the precipitation process is strongly affected by thermomechanical loads from the processing process and we accurately quantify these. Specifically, this research leads to the development of new spall resistant magnesium alloys, suited for extreme environments.
2. Coarse grained and linear scaling Density Functional Theory: Defects in materials
Defects in crystalline solids, play crucial role in determining macroscopic properties of materials. The profound significance of defects underlies from the coupling between the discrete effects of the lattice, chemical effects of the core and the long-range effects of the elastic field. While Density Functional Theory (DFT) is capable of accurately describing the chemistry of the defect core, but are too complicated and expensive for defects with long range fields, methods capable for describing long-range fields rely on empiricism and lack fidelity. This poses an outstanding dual challenge to simulate defects from first principles. To overcome this, we develop a novel coarse-grained formulation of DFT. We employ Linear Scaling Spectral Quadrature method to solve for the electronic fields and develop a coarse-graining strategy based on updated Lagrangian method to describe the long-range fields. The developed real space formulation is based on high-order finite differences, local reformulation of electrostatics, reformulation of the atomic forces and a parallelization strategy based on domain decomposition method. The developed formulation is sublinear scaling with respect to the system size and can efficiently simulate defects in crystalline materials from first principles.
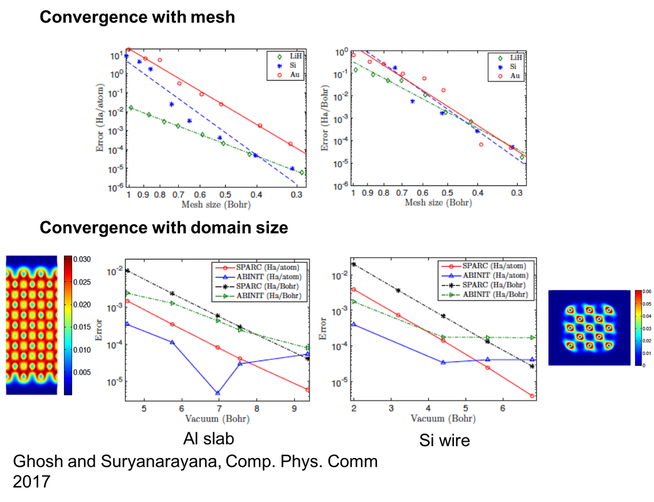
3. Large-scale real-space electronic structure methods
Density Functional Theory (DFT) is a widely used and popular electronic structure theory. The plane-wave basis is commonly employed for solving the DFT problem. However, the need for periodicity limits the effectiveness of the plane-wave basis in studying localized or partially periodic systems. Furthermore, efficient utilization of modern large-scale computer architectures is particularly challenging due to the non-locality of the basis. Real-space methods for solving the DFT problem provide an attractive alternative. In this research we develop an accurate and efficient real-space formulation and parallel implementation of Density Functional Theory (DFT) for performing ab-initio simulations of isolated clusters (molecules and nanostructures), periodic (infinite crystals) and partially periodic systems (slabs and nanowires). Using the finite-difference representation, local reformulation of the electrostatics, the Chebyshev polynomial filtered self-consistent field iteration, and a reformulation of the non-local component of the force, we develop SPARC (Simulation Package for Ab-initio Real-space Calculations), a framework that enables the efficient evaluation of energies and atomic forces to within chemical accuracies in DFT. Through selected examples consisting of a variety of elements, we demonstrate several attractive features of the developed framework. Overall, the developed framework is able to accurately and efficiently simulate the electronic structure of a wide range of material systems and represents an attractive alternative to existing codes for practical DFT simulations.
4. Mechanics of nanomaterials: Bending deformations and symmetry reduction
Nanostructures with coupled electro-mechanical and thermo-mechanical properties can be exploited for developing wearable devices, flexible electronics, drug delivery systems, energy harvesters, sensors and actuators. These systems are likely to be under large strains and often under bending deformations. The state of the art methods for analyzing these nanotubes and nanostructures under bending deformations are restricted by the size of systems that can be studied, or by the use semi-empirical theories (e.g. Tight Binding) that cannot accurately quantify electronic and thermal properties. We have overcome this by incorporating ideas of symmetry into ab-initio methods. To this end, I have extended SPARC to incorporate cyclic symmetry, which allows using first principles theory for studying the influence of bending deformations on nanostructures and to simulate very large nanotubes. By using ideas of objective structures, we have recently used this framework to simulate a nanotube with 1000 nm radius. This is the first time a simulation of a micron scale nanotube has been performed solely from first principles.
SCIENTIFIC SOFTWARE DEVELOPED
SPARC
Cylindrical-DFT
MacroDFT